


Resources

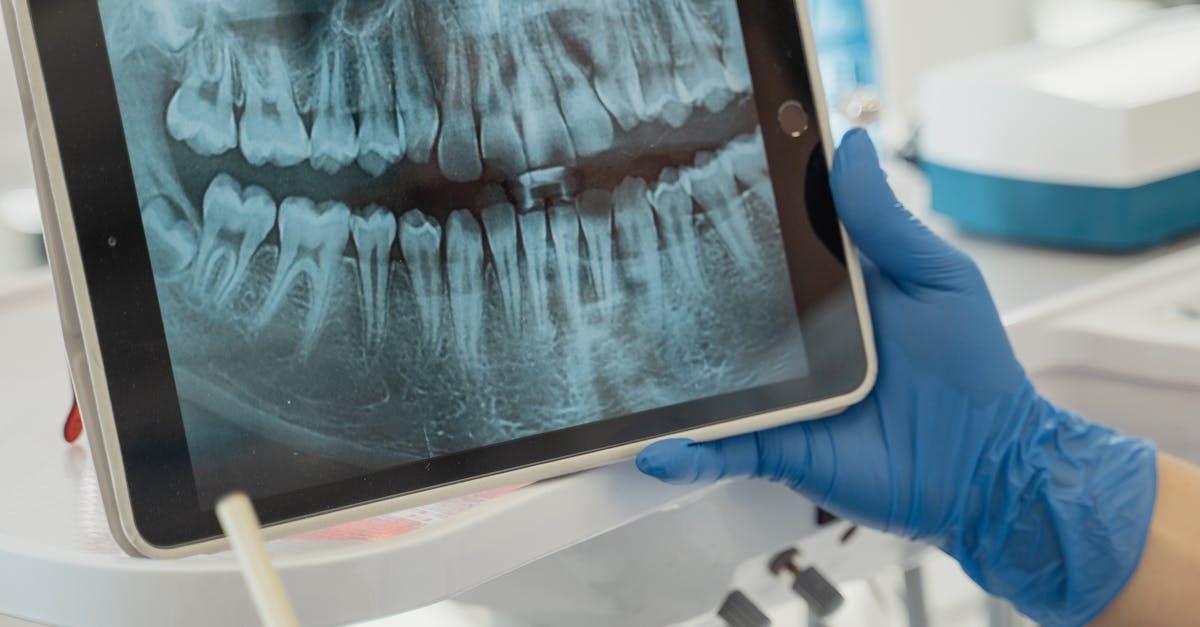
If you are a U.S.-based manufacturer of Software as a Medical Device (SaMD) looking to commercialize your products in Europe, obtaining CE marking is a mandatory step. Compliance with the Medical Device Regulation (EU) 2017/745 (MDR) ensures that your software meets the required safety, performance, and regulatory standards for legal marketing in Europe. Below, we outline the key steps to achieve CE marking certification. Step 1: Implement a Quality Management System (QMS) – ISO 13485 Certification To comply with MDR, manufacturers must implement a Quality Management System (QMS) aligned with ISO 13485. This standard ensures that medical devices are consistently designed, developed, and maintained in accordance with regulatory requirements. For U.S.-based companies, the FDA’s Quality System Regulation (QSR) (21 CFR Part 820) is being updated to align more closely with ISO 13485, with the Quality Management System Regulation (QMSR) taking effect in 2026. However, compliance with ISO 13485 is mandatory for CE marking in Europe. Step 2: Determine Your SaMD classification under MDR 2017/745 and MDCG 2019-11 Rule 11 (Annex VII, chapter III) of the MDR 2017/745 determines the classification of SaMD based on their intended use and potential risks. Class I (Low-risk) – Software with no clinical decision-making role. Class IIa (Moderate risk) : Software that provides medical information used for diagnostic or therapeutic purposes but whose incorrect use is unlikely to cause serious harm. Class IIb (high risk) : Software that provides medical information that could result in serious health deterioration or require surgical intervention in the event of failure or incorrect use. Class III (critical risk) : Software that directly drives or influences medical decisions, where failure could result in death or irreversible health deterioration. Step 3: Prepare your Technical Documentation for CE Marking Your technical file must comply with MDR and includes the following essential documents: Device description, including qualification and classification according to MDR Rule 11. Software development lifecycle documentation (compliant with IEC 62304, IEC 82304). Clinical evaluation report and risk management (compliant with ISO 14971) demonstrating safety and performance. Post-market surveillance (PMS) plan and vigilance reporting system, including periodic safety update reports (PSURs). Labeling and instructions for use (IFU). Declaration of conformity. Usability engineering file (compliant with IEC 62366). UDI (unique Device identification) (see our article on UDI system ). Verification and validation reports. this is a non-exhaustive list, there are other documents that constitute the Technical File. Step 4: Choose a Notified Body (if applicable) For Class I medical devices, must self-certify compliance with MDR but are still required to register their device with the Competent Authority in the EU member state where their Authorized Representative (AR) is established. For Class IIa, IIb, or III SaMD, a Notified Body (NB) must validate MDR compliance by delivering the CE-marking certificate. Step 5: Register Your Company and Product on EUDAMED Under MDR, all manufacturers must register in EUDAMED, the European Database on Medical Devices, which centralizes information about economic operators, devices, and post-market surveillance. EUDAMED is designed to enhance transparency and traceability within the EU market. You and your Authorized Representative must both register in the Economic Operators module. Each device must be registered in the UDI/Device Registration module, ensuring compliance with MDR Article 27 on traceability. Conclusion For a U.S.-based SaMD company, entering the European market requires strict adherence to MDR 2017/745. The key steps include: Implementing ISO 13485 QMS. Classifying the SaMD under MDR. Preparing complete technical documentation. Obtaining CE certification (if required). Registering in EUDAMED. With the right regulatory approach, your software can successfully enter the European market.

In our previous article, we explored the key steps for US manufacturers of SaMD to obtain CE marking and enter the European market (see our article : “ How to Market Your Medical Device in Europe?”) . One of the requirements of the Medical Device Regulation (EU) 2017/745 (MDR) is the appointment of an Authorized Representative (AR). Indeed, non-European manufacturers must appoint an AR to liaise between the manufacturer and the European competent authorities. This role is essential to ensure compliance, manage regulatory obligations and facilitate market access. In this article, we examine the importance of appointing an authorized representative. Why Is an Authorized Representative Mandatory ? According to Article 11 of MDR, the AR serves as the legal representative of a non-EU manufacturer within the EU and carries several responsibilities: Verifying the Declaration of Conformity and the conformity of the Technical Documentation. Holding a copy of the Technical Documentation and make it available to Competent authorities. Cooperating with the Competent Authorities. Informing the manufacturer in case of an accident. ➡️ Without an AR, your software cannot be marketed in Europe. The AR is not the only one with responsibilities, the manufacturer (Non-EU Company) holds following obligations: Ensuring that the medical device complies with MDR requirements before placing it on the market. Establishing and maintaining a Technical Documentation in accordance with MDR. Implementing a Quality Management System (QMS) (compliant with the standard ISO 13485). Conducting clinical evaluations (both pre-market and post-market). Ensuring compliance with post-market surveillance (PMS) requirements. Registering the manufacturer and its devices in EUDAMED and ensuring UDI compliance. Conclusion Entering the European market with a SaMD starts with obtaining CE marking. Only then can U.S. manufacturers appoint an Authorized Representative (AR), as required by the Medical Device Regulation (EU) 2017/745 (MDR). The AR plays a vital role in ensuring compliance and facilitating market access.

Softwares as Medical Devices (SaMD) integrating Artificial Intelligence (AI) are becoming increasingly common in healthcare, which is good news for improving the efficiency of our healthcare system and enhancing patient care.

Digital therapeutics (DTx) are innovative digital medical devices designed to treat various pathologies and alleviate their symptoms. Unlike traditional medical devices, DTx achieve their therapeutic effects primarily through cognitive-behavioral modifications (like CBT) facilitated by digital platforms, rather than pharmacological or immunological means. Why Digital Therapeutics Need CE Marking DTx have medical purposes and, as such, fall under the MDR definition of medical devices. Whether it’s helping patients manage chronic diseases or treating mental health diseases’ symptoms, DTx must demonstrate compliance with MDR requirements through the CE marking process. This involves robust technical documentation, clinical evaluations, and quality management practices (implementation of a Quality Management System). Classification of Digital Therapeutics (DTx) Under Rule 11 of the MDR Rule 11 of the MDR 2017/745 determines the classification of Software as Medical Devices based on their intended use and potential risks. DTx are generally classified as Class I, IIa, or IIb, depending on the complexity of their functionalities and their impact on patient health. The regulatory pathway varies according to the classification. For Class I medical devices, self-certification is sufficient to obtain CE marking. For higher classes, manufacturers must certify their devices with a Notified Body. Therefore, it is crucial to justify the chosen classification in detail, relying not only on Rule 11 of the MDR 2017/745 but also on the MDCG 2019-11 guidance on the qualification and classification of software. This document provides additional guidelines for evaluating whether a software qualifies as a medical device and for rigorously determining its risk class. Below are specific examples relevant to DTx: Class I (Low Risk): DTx tools offering basic functionalities without directly influencing medical decisions. Example: Software that monitors general health parameters (such as symptoms or physical condition) and provides a summary or personalized tracking, without clinical interpretation of the data, therapeutic recommendations, or alerts for healthcare professionals. Class IIa (Moderate Risk): DTx devices that provide information to assist in clinical decision-making, but where the consequences of failure are considered low. Example: An app that assists in diagnosing hearing dysfunction by interpreting auditory test data and proposes a personalized program to manage symptoms, while leaving the final decision to the healthcare professional. Class IIb (High Risk): DTx devices that provide critical information for diagnostic or therapeutic decision-making, where failure could result in serious harm to the patient. Strategic Considerations for DTx Manufacturers For DTx manufacturers, determining the appropriate classification is a critical step in balancing speed to market with regulatory compliance. While Class I devices with limited functionalities benefit from a simpler CE marking process, technical documentation remains essential to demonstrate compliance with the general safety and performance requirements. Conversely, for Class IIa or IIb devices, which are often associated with more complex therapies, manufacturers must prepare more comprehensive technical documentation and provide robust clinical validation data to meet stricter regulatory requirements. Conclusion The CE marking process is essential for digital therapeutics, ensuring they meet high standards of safety and effectiveness. By understanding the classification rules specific to DTx and aligning development strategies with MDR requirements, manufacturers can successfully bring their innovative solutions to the European market. Whether targeting basic self-management tools or advanced data-driven interventions, the classification of DTx under Rule 11 provides a clear regulatory framework to guide their development.
Published on July 12, Regulation 2024/1689, also known as the AI Act, establishes a comprehensive regulatory framework for AI, affecting all stakeholders, including those outside the European Union. Key Points to Remember 💡 Risk Classification: AI systems are now classified into four risk levels, ranging from unacceptable risk to minimal risk. High-risk systems, such as certain Medical Devices (MDs), will require specific measures, including human oversight and stringent technical standards. Conformity Requirements: For high-risk systems, implementing a Quality Management System (QMS) compliant with ISO 42001, developing technical documentation, and obtaining CE marking are mandatory. Crucial Deadline: Mark August 2, 2027, in your calendars, a critical date for the compliance of high-risk MDs with the AI Act. Next Steps to Consider ➡️ Review the Complete Text of the AI Act to understand its implications for your organization. Evaluate Your AI Systems to determine their risk classification under the new regulation. Develop a Global Regulatory Strategy that integrates the General Data Protection Regulation (GDPR), the Medical Device Regulation (MDR 2017/745), and the In Vitro Diagnostic Regulation (IVDR 2017/746) to ensure comprehensive regulatory compliance. Engage a Notified Body to assess the AI aspect of your systems. Adapt Your Compliance Processes to meet the specific requirements for your AI systems. Prepare now to navigate this new regulatory era successfully!

A question we often encounter is whether it is possible to start with self-certification for a Class I Medical Device (MD) and then upgrade it to a Class IIa. Let's delve into the topic: What is the difference in classification? Class I includes, for example, MDs like Digital Therapeutics (DTx) for patient use, whereas Class IIa encompasses more complex MDs, such as diagnostic support software or telemonitoring software that includes alerts for healthcare professionals. The rationale behind the classification of an MD primarily depends on its intended use and potential risks to patients. Class I MDs are generally self-managed by manufacturers through self-certification, significantly speeding up their market entry. In some cases, it can be very advantageous for manufacturers to start with a Class I software MD to reach the market and simultaneously carry out their clinical, regulatory, and product strategy to develop and certify a Class IIa MD. Class IIa MDs undergo stricter regulatory controls due to their complexity and increased risk potential, including audits by Notified Bodies (NB). What needs to be done to update the Medical Device class? Manufacturers considering upgrading their Class I MDs to Class IIa must prepare for a substantial update to their CE marking: Technical Documentation and Quality Management System (QMS). Any significant changes in the design or intended use of the device that could influence its class must be rigorously documented. Clinical evidence must be strengthened. From a financial perspective, designing a Class IIa MD is more expensive. Indeed, a Notified Body must be approached to evaluate the MD and issue the CE marking. Schedule an appointment with the Sparta Care team to assess the risk class of your MD and the best marking strategy to adopt!

The new Regulation (EU) 2017/745 of the European Parliament and the Council of April 5, 2017, on medical devices, requires more rigorous traceability of medical devices to increase manufacturer transparency towards patients. This requirement has led to the creation of the UDI system. The UDI system consists of several codes that manufacturers will need to enter into a new public database: EUDAMED. This system can be difficult to grasp, which is why we propose to review together the different parts of the UDI: UDI: Unique Device Identification It is represented by a barcode consisting of a static part (UDI-DI) and a dynamic part (UDI-PI). UDI-PI: A code that identifies the production of the medical device (e.g., lot number, release date, expiration date…) and thus represents the dynamic part of the UDI. UDI-DI: A code that uniquely identifies the medical device (the specific model!) and thus represents the static part of the UDI. Basic UDI-DI: A code that identifies a group of similar medical devices with variations that do not impact their basic application. Example: A remote monitoring software medical device with several variants (one variant per pathology) will have a unique Basic UDI-DI. However, each variant will have its own UDI-DI. 💡 To obtain the UDI-DI and Basic UDI-DI codes, it is necessary to contact authorized international coding bodies, such as GS1. The assignment of the Basic UDI-DI is free and can be done autonomously on the GS1 website. However, the UDI-DI is a paid service that can only be obtained after joining GS1. It is also important to note that in addition to the GS1 subscription, an additional cost is charged for each product UDI-DI requested.

D-30 before one of the deadlines of the MDR 2017/745 transition period: May 26, 2024.
Initiate an evaluation request with a Notified Body.
Ensure the compliance of your QMS.

A significant portion of the compliance costs in the Medical Device (MD) sector could be avoided. For more than 40% of companies, these regulatory costs are estimated to exceed 500,000 euros. Nearly 50% of these costs are actually related to what I identify as regulatory drift, often due to the lack of a clearly defined roadmap.
Obtaining the CE marking has become a real race against time. Currently, with the transition period of the European Regulation 2017/745 on Medical Devices (MDR) and the overwhelmed Notified Bodies (NBs), the time to obtain this marking extends between 12 and 18 months.